


Come usare LUXTURNA
Tradotto con IA
Questa pagina fornisce informazioni generali e non sostituisce la consultazione di un medico. Consulta sempre un medico prima di assumere qualsiasi medicinale. Rivolgiti ai servizi di emergenza se i sintomi sono gravi.
Mostra traduzioneContenuto del foglietto illustrativo
Luxturna 5 × 10 genomi vettoriali/mL concentrato e solvente per soluzione iniettabile
voretigene neparvovec
Medicinale sottoposto a monitoraggio addizionale. Ciò permetterà la rapida identificazione di
nuove informazioni sulla sicurezza. Lei può contribuire segnalando qualsiasi effetto indesiderato
riscontrato durante l’assunzione di questo medicinale. Vedere la fine del paragrafo 4 per le
informazioni su come segnalare gli effetti indesiderati.
Legga attentamente questo foglio prima che le sia somministrato questo medicinale perchè
contiene importanti informazioni per lei.
- Conservi questo foglio. Potrebbe aver bisogno di leggerlo di nuovo.
- Se ha qualsiasi dubbio, si rivolga al medico o all’infermiere.
- Se si manifesta un qualsiasi effetto indesiderato, compresi quelli non elencati in questo foglio, si rivolga al medico o all’infermiere. Vedere paragrafo 4.
Contenuto di questo foglio
- 1. Cos’è Luxturna e a cosa serve
- 2. Cosa deve sapere prima che le venga somministrato Luxturna
- 3. Com’è somministrato Luxturna
- 4. Possibili effetti indesiderati
- 5. Come conservare Luxturna
- 6. Contenuto della confezione e altre informazioni
1. Cos’è Luxturna e a cosa serve
Luxturna è un prodotto di terapia genica che contiene il principio attivo voretigene neparvovec.
Luxturna è utilizzato per il trattamento di adulti e bambini con perdita della vista dovuta a distrofia
retinica ereditaria causata da mutazioni nel gene RPE65. Queste mutazioni impediscono di produrre
una proteina necessaria per la visione e quindi portano alla perdita della vista e conseguente cecità.
Il principio attivo contenuto in Luxturna, voretigene neparvovec, è un virus modificato che contiene
una copia funzionante del gene RPE65. Dopo iniezione, rilascia questo gene nelle cellule della retina,
lo strato posteriore dell’occhio che rileva la luce. Ciò consente alla retina di produrre le proteine
necessarie per la visione. Il virus utilizzato per rilasciare il gene non causa malattie negli esseri umani.
Luxturna le verrà dato solo se test genetici mostrano che la sua perdita della visione è causata dalla
mutazione del gene RPE65.
2. Cosa deve sapere prima che le venga somministrato Luxturna
Non le deve essere somministrato Luxturna
- se è allergico a voretigene neparvovec o ad uno qualsiasi degli altri componenti di questo medicinale (elencati al paragrafo 6)
- se ha un’infezione agli occhi
- se ha un’infiammazione agli occhi
Se è presente una qualsiasi delle condizioni sopra descritte, o se non è sicuro di quanto sopra, è
pregato di parlare con il medico prima di ricevere Luxturna.
Avvertenze e precauzioni
Prima di ricevere il trattamento con Luxturna:
- Informi il medico se ha segni di un'infezione agli occhi o un'infiammazione agli occhi, ad esempio se ha arrossamento degli occhi, sensibilità alla luce, gonfiore oculare o dolore agli occhi.
- Informi il medico se ha un’infezione attiva di qualsiasi tipo. Il medico può ritardare il trattamento fino alla scomparsa dell’infezione poiché questo medicinale può rendere più difficile debellare un’infezione. Vedere anche il paragrafo 3.
Dopo aver ricevuto Luxturna:
- Contatti immediatamente il medico se il suo occhio o occhi diventano rossi, dolenti, sensibili alla luce, se vede lampi o corpi mobili, o se nota un peggioramento o annebbiamento della vista.
- Deve evitare viaggi aerei o altri viaggi ad altitudini elevate fino a quando consigliato dal medico. Durante il trattamento con questo medicinale, il medico introduce una bolla d'aria nell'occhio, che viene assorbita lentamente dal corpo. Fino a quando la bolla non viene completamente assorbita, viaggi aerei o altri viaggi a quote elevate possono fare espandere la bolla e determinare danni agli occhi, inclusa la perdita della vista. Si prega di parlare con il medico prima di viaggiare.
- Deve evitare di nuotare a causa di un aumentato rischio di infezione negli occhi. Si prega di parlare con il medico prima di andare a nuotare dopo aver ricevuto il trattamento con Luxturna.
- Deve evitare l'attività fisica intensa a causa di un aumentato rischio di lesioni agli occhi. Si prega di parlare con il medico prima di iniziare una intensa attività fisica dopo aver ricevuto Luxturna.
- Può accusare disturbi visivi temporanei, come sensibilità alla luce e visione offuscata. Informi il medico di eventuali disturbi visivi che si verificano. Il medico può essere in grado di contribuire alla riduzione di qualsiasi disagio causato da questi disturbi temporanei.
- Il principio attivo di Luxturna può essere temporaneamente escreto attraverso le lacrime. Lei e chi la segue dovete riporre le medicazioni usate e il materiale di scarto con lacrime e secrezioni nasali in sacchetti sigillati prima dello smaltimento. Dovrete seguire queste precauzioni per 14 giorni.
- Potrebbe non essere in grado di donare sangue, organi, tessuti e cellule per il trapianto dopo il trattamento con Luxturna.
Bambini e adolescenti
Luxturna non è stato studiato in bambini di età inferiore a quattro anni. I dati sono limitati.
Altri medicinali e Luxturna
Informi il medico se sta usando, ha recentemente usato o potrebbe usare qualsiasi altro medicinale.
Gravidanza, allattamento e fertilità
Se è in corso una gravidanza, se sospetta o sta pianificando una gravidanza o se sta allattando con latte
materno chieda consiglio al medico o all’infermiere prima che le sia somministrato Luxturna.
Gli effetti di questo medicinale sulla gravidanza e sul nascituro non sono noti. Come precauzione, non
deve ricevere Luxturna mentre è incinta.
Luxturna non è stato studiato nelle donne che allattano con latte materno. Non è noto se passi nel latte
materno. Informi il medico se sta allattando al seno o ha in programma di farlo. Il medico la aiuterà
quindi a decidere se interrompere l'allattamento al seno o non ricevere Luxturna, tenendo conto del
beneficio dell'allattamento al seno per il suo bambino e del beneficio di Luxturna per lei.
Guida di veicoli e utilizzo di macchinari
Dopo trattamento con Luxturna può avere disturbi visivi temporanei. Non guidi o non usi macchinari
pesanti fino a quando la vista non si è ripresa. Parli con il medico prima di riprendere queste attività.
Luxturna contiene sodio
Questo medicinale contiene meno di 1 mmol di sodio (23 mg) per dose, cioè essenzialmente ‘privo di
sodio’.
3. Com’è somministrato Luxturna
Luxturna le verrà somministrato in sala operatoria da chirurghi esperti in chirurgia oculare.
Luxturna viene somministrato in anestesia. Il medico le parlerà dell’anestesia e di come le verrà
somministrata.
Il medico eseguirà un intervento chirurgico oculare per rimuovere il gel trasparente all'interno
dell'occhio, e poi inietterà Luxturna direttamente sotto la retina, lo strato sottile sensibile alla luce nella
parte posteriore dell'occhio. La procedura verrà ripetuta nell’altro occhio almeno 6 giorni dopo. Dopo
ciascuna procedura, dovrà rimanere in osservazione post-operatoria per qualche ora per monitorare il
recupero e per osservare eventuali effetti indesiderati dovuti all'intervento chirurgico o all'anestesia.
Prima che inizi il trattamento con Luxturna il medico può chiederle di assumere un farmaco che
sopprimerà il suo sistema immunitario (le difese naturali del corpo) in modo che quest’ultimo non
neutralizzi Luxturna quando le viene dato. È importante prendere questo medicinale secondo le
istruzioni fornite. Non smetta di prendere il farmaco senza prima parlare con il medico.
Se le viene somministrato più Luxturna di quanto dovrebbe
Poiché questo medicinale le è stato dato da un medico, è improbabile che gliene venga dato troppo. Se
ciò accade, il medico tratterà i sintomi secondo necessità. Informi il medico o l'infermiere in caso di
problemi visivi.
Se ha qualsiasi dubbio sull’uso di questo medicinale, si rivolga al medico o all’infermiere.
4. Possibili effetti indesiderati
Come tutti i medicinali, questo medicinale può causare effetti indesiderati sebbene non tutte le persone
li manifestino.
I seguenti effetti indesiderati possono verificarsi con Luxturna:
Comuni (possono interessare fino a 1 persona su 10)
- Depositi sotto la retina
Non noti (la frequenza non può essere definita sulla base dei dati disponibili)
- Atrofia della (corio)retina
I seguenti effetti indesiderati possono verificarsi con la procedura di iniezione:
Molto comuni (possono interessare più di 1 persona su 10)
- Arrossamento degli occhi
- Cataratta (opacità del cristallino)
- Aumento della pressione all’interno dell’occhio
Comuni (possono interessare fino a 1 persona su 10)
- Rottura nella retina
- Dolore degli occhi
- Gonfiore dell’occhio
- Distacco della retina
- Sanguinamento nella parte posteriore dell’occhio
- Dolore o aumento del fastidio oculare
- Offuscamento della visione centrale dovuto a un foro al centro della retina
- Assottigliamento della superficie dell’occhio (dellen)
- Irritazione oculare
- Infiammazione oculare
- Sensazione di corpo estraneo nell’occhio
- Fastidio oculare
- Anomalie nella parte posteriore dell’occhio
- Nausea (malessere), vomito, dolore addominale (alla pancia), dolore al labbro
- Cambiamento dell’attività elettrica del cuore
- Mal di testa, capogiro
- Eruzione cutanea, gonfiore del viso
- Ansia
- Problemi associati con il posizionamento di un tubo per la respirazione nella trachea
- Rottura della ferita chirurgica
Non noti (la frequenza non può essere definita sulla base dei dati disponibili)
- Appannamento nella sostanza gelatinosa all’interno dell’occhio (opacità vitreali)
- Atrofia della (corio)retina
Un danno ai tessuti dell'occhio può essere accompagnato da sanguinamento e gonfiore e da un
aumento del rischio di infezione. Nei giorni successivi all'intervento la visione è ridotta e solitamente
migliora; informi il medico se la visione non ritorna.
Segnalazione degli effetti indesiderati
Se manifesta un qualsiasi effetto indesiderato, compresi quelli non elencati in questo foglio, si rivolga
al medico o all’infermiere. Può inoltre segnalare gli effetti indesiderati direttamente tramite il sistema
nazionale di segnalazione riportato nell’ allegato V . Segnalando gli effetti indesiderati può contribuire
a fornire maggiori informazioni sulla sicurezza di questo medicinale.
5. Come conservare Luxturna
Luxturna sarà conservato dagli operatori sanitari presso la sua struttura sanitaria.
Il concentrato e il solvente devono essere conservati e trasportati congelati a ≤-65 ºC. Una volta
scongelato, il medicinale non deve essere nuovamente congelato e deve essere lasciato a temperatura
ambiente (inferiore a 25 °C).
Non usi questo medicinale dopo la data di scadenza che è riportata sull’etichetta e sulla scatola dopo
EXP e Scad, rispettivamente.
6. Contenuto della confezione e altre informazioni
Cosa contiene Luxturna
- Il principio attivo è voretigene neparvovec. Ogni mL di concentrato contiene 5 × 10 genomi vettoriali ( vector genomes, vg). Il concentrato (0,5 mL di volume estraibile in un flaconcino monodose da 2 mL) richiede una diluizione 1:10 prima della somministrazione.
- Ogni dose di soluzione diluita contiene 1,5 × 10 genomi vettoriali di voretigene neparvovec in un volume erogabile di 0,3 mL.
- Gli altri componenti del concentrato sono sodio cloruro (vedere “Luxturna contiene sodio” al paragrafo 2 di questo foglio), sodio diidrogeno fosfato monoidrato (per l’aggiustamento del pH), disodio idrogeno fosfato diidrato (per l’aggiustamento del pH), polassamero 188 e acqua per preparazioni iniettabili.
- Il solvente contiene sodio cloruro (vedere la fine del paragrafo 2), sodio diidrogeno fosfato monoidrato (per l’aggiustamento del pH), disodio idrogeno fosfato diidrato (per l’aggiustamento del pH), polassamero 188 e acqua per preparazioni iniettabili.
Questo medicinale contiene organismi geneticamente modificati.
Descrizione dell’aspetto di Luxturna e contenuto della confezione
Luxturna è un concentrato per soluzione per iniezione sottoretinica limpido, incolore, fornito in un
flaconcino di plastica trasparente. Il solvente è un liquido limpido incolore fornito in un flaconcino di
plastica trasparente.
Ogni imballaggio di alluminio contiene un contenitore che racchiude 1 flaconcino da 0,5 mL di
concentrato e 2 flaconcini di solvente (ciascuno contente 1,7 mL).
Titolare dell’autorizzazione all’immissione in commercio
Novartis Europharm Limited
Vista Building
Elm Park, Merrion Road
Dublin 4
Irlanda
Produttore
Novartis Pharma GmbH
Roonstrasse 25
90429 Norimberga
Germania
Per ulteriori informazioni su questo medicinale, contatti il rappresentante locale del titolare
dell’autorizzazione all’immissione in commercio:
België/Belgique/Belgien
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Lietuva
SIA Novartis Baltics Lietuvos filialas
Tel: +370 5 269 16 50
България
Novartis Bulgaria EOOD
Тел: +359 2 489 98 28
Luxembourg/Luxemburg
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Česká republika
Novartis s.r.o.
Tel: +420 225 775 111
Magyarország
Novartis Hungária Kft.
Tel.: +36 1 457 65 00
Danmark
Novartis Healthcare A/S
Tlf: +45 39 16 84 00
Malta
Novartis Pharma Services Inc.
Tel: +356 2122 2872
Deutschland
Novartis Pharma GmbH
Tel: +49 911 273 0
Nederland
Novartis Pharma B.V.
Tel: +31 88 04 52 111
Eesti
SIA Novartis Baltics Eesti filiaal
Tel: +372 66 30 810
Norge
Novartis Norge AS
Tlf: +47 23 05 20 00
Ελλάδα
Novartis (Hellas) A.E.B.E.
Τηλ: +30 210 281 17 12
Österreich
Novartis Pharma GmbH
Tel: +43 1 86 6570
España
Novartis Farmacéutica, S.A.
Tel: +34 93 306 42 00
Polska
Novartis Poland Sp. z o.o.
Tel.: +48 22 375 4888
France
Novartis Pharma S.A.S.
Tél: +33 1 55 47 66 00
Portugal
Novartis Farma - Produtos Farmacêuticos, S.A.
Tel: +351 21 000 8600
Hrvatska
Novartis Hrvatska d.o.o.
Tel. +385 1 6274 220
România
Novartis Pharma Services Romania SRL
Tel: +40 21 31299 01
Ireland
Novartis Ireland Limited
Tel: +353 1 260 12 55
Slovenija
Novartis Pharma Services Inc.
Tel: +386 1 300 75 50
Ísland
Vistor hf.
Sími: +354 535 7000
Slovenská republika
Novartis Slovakia s.r.o.
Tel: +421 2 5542 5439
Italia
Novartis Farma S.p.A.
Tel: +39 02 96 54 1
Suomi/Finland
Novartis Finland Oy
Puh/Tel: +358 (0)10 6133 200
Κύπρος
Novartis Pharma Services Inc.
Τηλ: +357 22 690 690
Sverige
Novartis Sverige AB
Tel: +46 8 732 32 00
Latvija
SIA Novartis Baltics
Tel: +371 67 887 070
United Kingdom (Northern Ireland)
Novartis Ireland Limited
Tel: +44 1276 698370
Altre fonti d’informazioni
Questo foglio è disponibile come file audio e in larga stampa dal sito web:
http://www.voretigeneneparvovec.support
Informazioni più dettagliate su questo medicinale sono disponibili sul sito web dell’Agenzia europea
per i medicinali, http://www.ema.europa.eu .
------------------------------------------------------------------------------------------------------------------------
Le informazioni seguenti sono destinate esclusivamente agli operatori sanitari:
Precauzioni che devono essere prese prima della manipolazione o della somministrazione del
medicinale
Questo medicinale contiene organismi geneticamente modificati. Durante la manipolazione o la
somministrazione di voretigene neparvovec devono essere indossati dispositivi di protezione
individuale (inclusi camice da laboratorio, occhiali di sicurezza e guanti).
La pressione intraoculare deve essere monitorata prima e dopo la somministrazione del medicinale e
gestita in modo appropriato.
Dopo la somministrazione, i pazienti devono essere istruiti in modo tale da riportare senza ritardi ogni
sintomo indicativo di endoftalmite o distacco retinico e devono essere gestiti in modo appropriato.
Preparazione prima della somministrazione
Ciascuna confezione contiene 1 flaconcino di concentrato e 2 flaconcini di solvente monouso.
Luxturna deve essere ispezionato visivamente prima della somministrazione. Se si osservano
particolato, torbidità o scolorimento, il flaconcino monodose non deve essere usato.
La preparazione di Luxturna deve essere eseguita entro 4 ore dall'inizio della procedura di
somministrazione, in conformità con le seguenti raccomandazioni eseguite in condizioni asettiche.
Scongelare un flaconcino monodose di concentrato e due fiale di solvente a temperatura ambiente.
Una volta scongelati tutti e 3 i flaconcini (1 flaconcino di concentrato e 2 flaconcini di diluente), si
deve iniziare la diluizione. Capovolgere delicatamente i flaconcini cinque volte per mescolare il
contenuto.
Ispezionare per la presenza di eventuali particelle visibili o anomalie. Eventuali anomalie o particolato
visibile devono essere segnalati al titolare dell'autorizzazione all'immissione in commercio e il
prodotto non deve essere utilizzato.
Trasferire 2,7 mL del solvente estratto dai due flaconcini scongelati ed erogarli in un flaconcino sterile
vuoto di vetro da 10 mL utilizzando una siringa da 3 mL.
Per la diluizione, aspirare 0,3 mL di concentrato scongelato in una siringa da 1 mL e aggiungerlo al
flaconcino sterile da 10 mL contenente il solvente. Capovolgere delicatamente il flaconcino almeno
cinque volte per una corretta miscelazione. Ispezionare per la presenza di eventuale particolato
visibile. La soluzione diluita deve essere da limpida a leggermente opalescente. Etichettare il
flaconcino di vetro da 10 mL contenente il concentrato diluito come segue: "Luxturna diluito".
Non preparare le siringhe se il flaconcino presenta danni o se si osserva particolato visibile. Preparare
le siringhe per l'iniezione prelevando 0,8 mL di soluzione diluita in una siringa sterile da 1 mL.
Ripetere la stessa procedura per preparare una siringa di backup. Le siringhe riempite con il prodotto
devono quindi essere trasferite alla sala operatoria in un contenitore designato al trasporto.
Misure da adottare in caso di esposizione accidentale
L’esposizione accidentale deve essere evitata. Devono essere seguite le linee guida locali di
biosicurezza per la preparazione, la somministrazione e la manipolazione di voretigene neparvovec.
- Durante la manipolazione o la somministrazione di voretigene neparvovec devono essere indossati dispositivi di protezione individuale (tra cui camice da laboratorio, occhiali protettivi e guanti).
- L’esposizione accidentale a voretigene neparvovec, incluso il contatto con la pelle, occhi e mucose deve essere evitata. Eventuali ferite aperte devono essere coperte prima della manipolazione.
- Tutti gli sversamenti di voretigene neparvovec devono essere trattati con un agente virucida come l’ipoclorito di sodio all’1% e tamponati con materiali assorbenti.
- Tutto il materiale che può essere entrato in contatto con voretigene neparvovec (ad esempio, flaconcino, siringa, ago, garza di cotone, guanti, maschere, o medicazioni) deve essere smaltito in conformità con le linee guida locali di biosicurezza.
Esposizione accidentale
- In caso di esposizione professionale accidentale (ad esempi, attraverso uno schizzo agli occhi o alle mucose), sciacquare con acqua pulita per almeno 5 minuti.
- In caso di esposizione di pelle tagliata o di lesioni da ago, pulire accuratamente l’area interessata con sapone e acqua e/o un disinfettante.
Precauzioni da adottare per lo smaltimento del medicinale
Questo medicinale contiene organismi geneticamente modificati. Il medicinale non utilizzato o il
materiale di scarto devono essere smaltiti in conformità alle linee guida locali per i rifiuti farmaceutici.
Posologia
Il trattamento deve essere iniziato e somministrato da un chirurgo della retina esperto in chirurgia
maculare.
I pazienti riceveranno una singola dose di 1,5 × 10 genomi vettoriali di voretigene neparvovec in
ciascun occhio. Ogni dose verrà somministrata nello spazio sottoretinico in un volume totale di
0,3 mL. La procedura di somministrazione individuale in ciascun occhio viene eseguita in giorni
separati entro un intervallo ravvicinato, ma a non meno di 6 giorni di distanza.
Regime immunomodulatore
Prima dell’inizio del regime immunomodulatore e prima della somministrazione di voretigene
neparvovec, il paziente deve essere controllato rispetto alla presenza dei sintomi di infezioni attive di
qualsiasi natura e, in tal caso, l’inizio del trattamento deve essere posticipato a dopo la guarigione del
paziente.
A partire da 3 giorni precedenti la somministrazione di voretigene neparvovec nel primo occhi, si
raccomanda di iniziare un regime immunomodulatore seguendo il programma riportato di seguito
(Tabella 1). L'inizio del regime immunomodulatore per il secondo occhio deve seguire lo stesso
programma e sostituire il completamento del regime immunomodulatore per il primo occhio.
Tabella 1Regime immunomodulatore pre- e post-operatorio per ciascun occhio
Popolazioni speciali
Anziani
La sicurezza e l’efficacia di voretigene neparvovec nei pazienti di età ≥65 anni non sono state stabilite.
I dati sono limitati. Tuttavia, non è necessario un aggiustamento della dose nei pazienti anziani.
Compromissione epatica e renale
La sicurezza e l’efficacia di voretigene neparvovec nei pazienti con compromissione epatica o renale
non sono state stabilite. Non è necessario un aggiustamento della dose in questi pazienti (vedere
paragrafo 5.2).
Popolazione pediatrica
La sicurezza e l’efficacia di voretigene neparvovec nei bambini fino a 4 anni di età non sono state
stabilite. I dati sono limitati. Non è necessario un aggiustamento della dose nei pazienti pediatrici.
Modo di somministrazione
Uso sottoretinico.
Luxturna è una soluzione concentrata sterile per iniezione sottoretinica che richiede scongelamento e
diluizione prima della somministrazione.
Questo medicinale non deve essere somministrato tramite iniezione intravitreale.
Luxturna è un flaconcino monouso per una singola somministrazione in un solo occhio. Il prodotto
viene somministrato tramite iniezione sottoretinica dopo vitrectomia in ciascun occhio. Non deve
essere somministrato nelle immediate vicinanze della fovea per mantenere l’integrità foveale.
La somministrazione di voretigene neparvovec deve essere eseguita in sala operatoria in condizioni
asettiche controllate. Prima della procedura deve essere somministrata al paziente una anestesia
adeguata. La pupilla dell’occhio da iniettare deve essere dilatata e un microbiocida ad ampio spettro
deve essere somministrato per via topica prima dell’intervento secondo la normale pratica medica.
| Pre-operatorio | 3 giorni prima della somministrazione di Luxturna | o Prednisone (o equivalente) 1 mg/kg/die (massimo 40 mg/die) |
| Post-operatorio | 4 giorni (incluso il giorno della somministrazione) | c Prednisone (o equivalente) a 1 mg/kg/die (massimo 40 mg/die) |
| Seguito da 5 giorni | m Prednisone (o equivalente) 0,5 mg/kg/die (massimo 20 mg/die) | |
| Seguito da 5 giorni di una dose a giorni alterni | Prednisone (o equivalente) r 0,5 mg/kg a giorni alterni (massimo 20 mg/die) |
Somministrazione
Per somministrare voretigene neparvovec ai pazienti seguire i passaggi sottostanti:
- Dopo diluizione Luxturna deve essere ispezionato visivamente prima della somministrazione. Se si osservano particolato, torbidità, o scolorimento, il prodotto non deve essere usato.
- Collegare la siringa contenente il prodotto diluito al tubo di estensione e alla cannula per iniezione sottoretinica. Il prodotto viene lentamente iniettato attraverso il tubo di estensione e la cannula per iniezione sottoretinica per eliminare eventuali bolle d’aria nel sistema.
- Il volume di prodotto disponibile per l'iniezione è confermato nella siringa, allineando l’estremità dello stantuffo con la linea che segna 0,3 mL.
- Dopo il completamento della vitrectomia, Luxturna viene somministrato mediante iniezione sottoretinica utilizzando una cannula per iniezione sottoretinica introdotta tramite pars plana.
- Sotto visualizzazione diretta, la punta della cannula per iniezione sottoretinica viene posta a contatto con la superficie retinica. Il sito di iniezione raccomandato deve trovarsi lungo l’arcata vascolare superiore, ad almeno 2 mm di distanza dal centro della fovea. Una piccola quantità di prodotto viene iniettata lentamente fino alla iniziale osservazione di una vescichetta sottoretinica, quindi il volume rimanente viene iniettato lentamente per un totale di 0,3 mL (Figura 1).
Figura 1Punta della cannula per iniezione sottoretinica posizionata all’interno del sito di
iniezione raccomandato (vista del chirurgo)
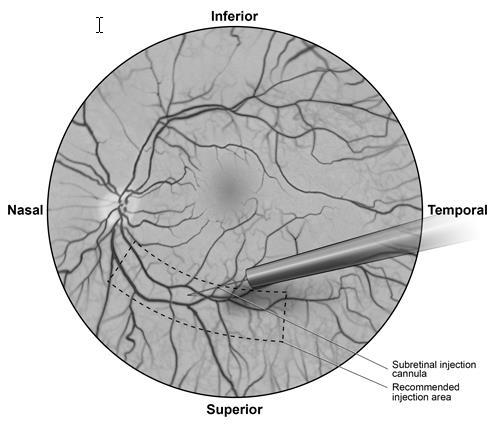
- Al termine dell’iniezione, la cannula per iniezione sottoretinica viene rimossa dall’occhio.
- Dopo l’iniezione, il prodotto inutilizzato deve essere eliminato. La siringa di back-up non può essere conservata.
- Viene effettuato uno scambio fluido-aria, evitando accuratamente il drenaggio dei liquidi vicino alla retinotomia effettuata per l’iniezione sottoretinica. Nel periodo post-operatorio, viene assunto immediatamente un posizionamento supino della testa che, in seguito alla dimissione, deve essere mantenuto dal paziente per 24 ore.
ALLEGATO IV
CONCLUSIONI SCIENTIFICHE E MOTIVAZIONI PER LA VARIAZIONE DEI
TERMINI DELL’AUTORIZZAZIONE ALL’IMMISSIONE IN COMMERCIO
Conclusioni scientifiche
Tenendo conto della valutazione del Comitato per la valutazione dei rischi in farmacovigilanza
( Pharmacovigilance and Risk Assessment Committee, PRAC) del Rapporto periodico di
aggiornamento sulla sicurezza ( Periodic Safety Update Report, PSUR) per voretigene neparvovec, le
conclusioni scientifiche del PRAC sono le seguenti:
Alla luce dei dati disponibili sull’atrofia corioretinica, il PRAC ha concluso che le informazioni sul
medicinale contenente voretigene neparvovec devono essere modificate di conseguenza.
Aggiornamento del paragrafo 4.8 del RCP per aggiungere la reazione avversa “atrofia corioretinica”
all'elenco di quelle correlate a voretigene neparvovec con frequenza “non nota” e ulteriori dettagli
aggiornato di conseguenza.
Avendo esaminato la raccomandazione del PRAC, il Comitato dei medicinali per uso umano
( Committee for Human Medicinal Products, CHMP) concorda con le relative conclusioni generali e
con le motivazioni della raccomandazione.
Motivazioni per la variazione dei termini dell’autorizzazione all’immissione in commercio
Sulla base delle conclusioni scientifiche su voretigene neparvovec il CHMP ritiene che il rapporto
beneficio/rischio del medicinale contenente voretigene neparvovec sia invariato fatte salve le
modifiche proposte alle informazioni sul medicinale.
Il CHMP raccomanda la variazione dei termini dell’autorizzazione all’immissione in commercio.


-

- Paese di registrazione
- Forma farmaceuticaSoluzione iniettabile, 5 X 10 ALLA DODICESIMA
- Codice ATCS01XA27
- Principio attivo
- Prescrizione richiestaSì
- Produttore
- Queste informazioni sono solo a scopo informativo e non costituiscono un parere medico. Consulta sempre un medico prima di assumere qualsiasi medicinale. Oladoctor non è responsabile delle decisioni mediche basate su questo contenuto.
- Alternative a LUXTURNAForma farmaceutica: Collirio, soluzione, 5%Principio attivo: acetylcysteineProduttore: BRUSCHETTINI S.R.L.Prescrizione non richiestaForma farmaceutica: Collirio, soluzione, 10MG/MLPrincipio attivo: artificial tears and other indifferent preparationsProduttore: ABBVIE S.R.L.Prescrizione non richiestaForma farmaceutica: Collirio, soluzione in contenitore monodose, 0,9 MG/MLPrincipio attivo: ciclosporinProduttore: SUN PHARMACEUTICAL INDUSTRIES (EUROPE) B.V.Prescrizione non richiesta
Medici online per LUXTURNA
Valutazione del dosaggio, effetti indesiderati, interazioni, controindicazioni e rinnovo della prescrizione di LUXTURNA — soggetto a valutazione medica e alle normative locali.







Ottieni una ricetta per LUXTURNA online
Compila un modulo di 2 minuti
Raccontaci i tuoi sintomi, la tua storia clinica e il medicinale che stai richiedendo.
Scegli un medico o lascia che lo assegniamo noi
Scegli uno specialista o ti abbineremo al primo medico disponibile.
Il medico esamina il tuo caso
Di solito entro 30 minuti. Potrebbe farti domande aggiuntive via chat.
Ritira la ricetta in qualsiasi farmacia
Ricetta elettronica inviata alla tua email — valida in tutta la Spagna.
Domande frequenti

Rimani aggiornato su Oladoctor
Novità su nuovi servizi, aggiornamenti del prodotto e contenuti utili per i pazienti.